Podsumowanie
Genetyka z Michigan XD
Podsumowanie
Genetyka z Michigan XD
Twój wynik
Wszystkie ({{dataStorage.userResults.answersTotal}})
Prawidłowe ({{dataStorage.userResults.answersGood}})
Do powtórki ({{dataStorage.userResults.answersRepeat}})
Błędne ({{dataStorage.userResults.answersBad}})
Pytanie 1
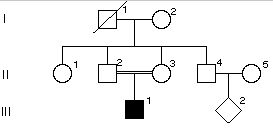
1. Dla danej choroby autosomalnej recesywnej q=0.01. Ile % populacji ma 2 kopie normalnego allelu?
2
98
95
19
90
Pytanie 2
2. Dziecko urodziło się z rozszczepieniem wargi i podniebienia. Może być to związane z:
a. schorzeniem w obrębie chromosomu np. 13
b. przerwaniem worka owodniowego
c. zdrowym, zupełnie normalnym dzieckiem
A i C
wszystkie powyzsz
Pytanie 3
3. Jeśli 1/250000 ma chorobę X, nieletalną, autosomalną recesywną, jaka jest częstość nosicielstwa?
1/250
1/25
1/1000
1/500
1/50
Pytanie 4
4. Linkage analysis (analiza odległości genów na chromosomie) została przeprowadzona w dużej rodzinie z autosomalną dominującą anemią hemolityczną, z użyciem polimorficznego markera z miejscem wiązania beta-globiny. Wynik LOD przy (theta)=0 to minus nieskończoność (?). LOD przy (theta)=0,01 to -4,5. Można wnioskować, że to zaburzenie w rodzinie:
c. to nabyte zaburzenie spowodowane mutacją w genie somatycznym
e. jest spowodowane mutacją w genie na chromosomie 11, 10cM centramerycznie od B-globuliny
b. nie jest spowodowane mutacją w genie beta-globiny
d. jest spowodowane mutacją w genie alfa-globiny
a. jest spowodowane mutacją w genie beta-globiny
Pytanie 5
5. Alkaptonuria jest spowodowana niedoborem:
c. kwasu homogentyzynowego
b. normalnie zabarwionego moczu
d. oksydazy fenyloalaninowej
a. hydroksylazy fenyloalaninowej
e. oksydazy kwasu homogentyzynowego
Pytanie 6
6. Przeobrażający się onkogen Simiain Sarcoma Virus (SSV) to sis gene. Przeprowadziłeś hybrydyzację Southerna na swoim własnym DNA używając sis gene jako sondy i znalazłeś kilka hybrydyzujących pasm. to świadczy o zakażeniu Twoich krwinek infekcją SSV
tak
nie
Pytanie 7
7. Częstość występowania autosomalnej dominującej rodzinnej hipercholesterolemii, wtórnej do heterozygotyczności (xD) dla mutacji LDL-R wynosi 1/500. 32 latek chory, hajtnął się z genetycznie niespokrewnioną 20-latką. Jakie jest prawdopodobieństwo, że ich dziecko będzie dotknięte ciężką formą tej choroby?
1/1000
1/250
1/2
1/2000
1/1000000
Pytanie 8
9. Wszystkie poniższe dają potwierdzający dowód na klonowe pochodzenie guza, OPROCZ:
a. U heterozygot wszystkie komorki guza wykazują zmniejszczenie względem homozygot
b. Wszystkie komorki guza posiadają dokładnie 46 chromosomow
d. Wszystkie komorki guza posiadają to samo przemieszczenie genów kodujących immunoglobulinę
c. Wszystkie komorki guza posiadają ten sam nienormalny chromosom
e. Guz od żeńskich heterozygot dla polimorfizmu G6PD. We wszystkich komórkach guza dochodzi do ekspresji tego samego allelu
Pytanie 9
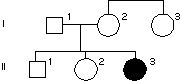
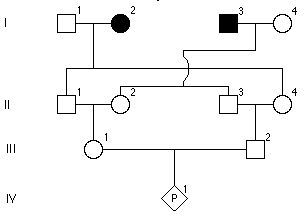
8.Rodowód rodziny z mukowiscydozą – chorobą autosomalną recesywną. Jakie jest prawdopodobieństwo, że I-3 jest nosicielem?
3/4
2/3
1/4
1/2
1/3
Pytanie 10
Genetyczne doradztwo musi się opierać na szanowaniu której z zasad etyki medycznej?
sprawiedliwości
opłacalności
autonomii
żadne z powyższych
paternalizmu
Pytanie 11
Gdy rodzice nie chcą się zgodzić na transfuzję krwi dziecka bo są Jehowcami to idziesz do sądu i powołujesz się na:
pufność
nieszkodzenie
sprawiedliwość
paternalizm
Pytanie 12
wynik pozytywny: A (dotknięty), B (niedotknięty) wynik negatywny: C (dotknięty), D (niedotknięty)
czułość
A/(A+C)
specyficzność
D/(D+B)
współczynnik fałszywej dodatności
B/(A+B)
współczynnik fałszywej ujemności:
C/(C+D)
Pytanie 13
FRB - siatkowczak rodzinny, jest czynnikiem autosomalnym dominującym predyspozycji do raka, z powodu defektowania genu supresorowego raka RB. Dotknięte jednostki najczęściej dziedziczą 1 defektową kopię genu od jednego rodzica i 1 zdrową od drugiego. Obrazek z Southerblotowanym materiałem z normalnych komorek i siatkowczaka. Co jest najbardziej prawdopodobnym wyjaśnieniem utraty heterozygotyczności?
niezależna, druga mutacja
mitotyczny crossover
utrata normalnego chromosomu 13 i replikacja zmutowanego chromosomu 13
utrata normalnego chromosomu 13
Pytanie 14
14.Analiza chromosomalna u młodej kobiety z łagodnymi objawami zespołu Turnera pokazała kariotyp 46,XX/45,X. Nondysjunkcja najprawdopodobniej zaszła w:
mitozie po zapłodnieniu
matczynej mejozie I
ojcowskiej mejozie II
matczynej mejozie II
ojcowskiej mejozie I
Pytanie 15
15.Co nie jest typowym mechanizmem przez ktory portoonkogen przemienia się w onkogen?
c.amplifikacja protoonkogenu
a.mutacja punktowa w protoonkogenie
b.całkowita delecja protoonkogenu
d.translokacja chromosomalna skutkujące zwiększeniem ekspresji portoonkogenu
Pytanie 16
Dopasuj:
MEN-II jest autosomalnym dominującym syndromem raka. Innym takim syndromem jest choroba von Hippel-Lindau ktorej gen znajduje się na chromosomie 3. Biorąc pod uwagę, że dają podobne objawy jest to przykład:
heterogeniczności locus
95% cierpiących na MEN-II ma raka komorek C tarczycy, i jakieśtam inne z tarczycą, 50% ma tego złego raka, 20% ma powiększoną taczycę czy coś takiego. Jest to przykład:
zrożnicowanej ekspresji
Ok. 10% guzow chromochłonnych (w rdzeniu nadnerczy) przejawia się u pacjentow w postaci podobnych objawow onkologicznych, jednym z ktorych jest mnoga gruczolakowatość wewnątrzwydzielnicza typu II (MEN-II). W większości MEN-II rodzin występują germlinowe mutacje w genie RET, znajdujące się na chromosomie 10. Najczęściej te mutacje są w jednej z wielu kodonow kodujących cysteinę. To przykład:
heterogeniczność alleliczna
Pytanie 17
17.Każde z poniższych jest mechanizmem skutkującym w aktywacji portoonkogenu oprocz:
c.przechwycenie sekwencji onkogenu przez retrowirusa
b.translokację chromosomalną łączącą fragmenty onkogenu i innego genu
a.amplifikacja onkogenu jako małe, mniejsze niż chromosom fragmenty
e.mutacja punktowa zmieniająca funkcję białkowego produktu onkogennego
d.inaktywacja onkogenu przez aktywność telomerazy
Pytanie 18
18.Dlaczego beta-thalasemia zazwyczaj jest dostrzegalna tylko zaraz po urodzeniu?
d.podjednostki Hb kodowane przez gen alfa są wystarczające tylko do urodzenia
e.produkt b-globulinowego pseudogenu podlega mocnej ekspresji tylko po urodzeniu
c.HbF pozostaje po urodzeniu, kiedy powinna być usunięta
a.zdrowe krwinki mamy dostarczają tlenu do płodu
b.zmiana ekspresji z gamma-genu do beta genu następuje w czasie urodzenie
Pytanie 19
c.mitochondrialna
e.sprzężona z X dominująca
d.autosomalna recesywna
b.sprzężona z X recesywna
a.autosomalna dominująca
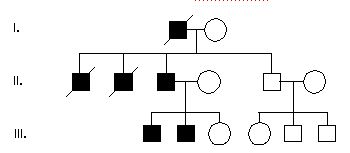
Pytanie 20
20.W rodowodzie poniżej wszyscy w I mają alfa-thalasemię. I-1 jest afrykańskiego pochodzenia. Dziecko II-2 umarło przez HF – obrzęk uogolniony płodu. Jaki najprawdopodobniej jest fenotyp II-1
e.tylko alfa thalasemia
b.HbH albo HF
d.cichy nosiciel albo HbH
c.normalny albo cichy nosiciel
a.normalny, alfa thalasemia albo HF
Pytanie 21
21.Choroba autosomalna recesywna występuje z częstością 1/10000 w populacji (zakładamy, że w danej miejscowości jest 100k ludu). Kumpel twierdzi, że ma chorobę chociaż nie miał w rodzinie bo test wyszedł pozytywnie (98% wrażliwości, 90% specyficzności). Jaka jest szansa, że jednak nie ma?
83%
49%
10%
17%
2%
Pytanie 22
22.Jakie jest dziedziczenie w tym rodowodzie?
a.autosomalne recesywne
c.sprzężone z X dominujące
d.sprzężone z X recesywne
b.autosomalne dominujące
e.mitochondrialne

Pytanie 23
23.Ktore z poniższych nie pomoże pacjentowi z beta-talasemią major (najcięższa)
e.wzrost produkcji HbF
c.spadek produkcji alfa-globuliny
b.wzrost produkcji alfa-globuliny
d.transfuzja i chelatacja żelazem
a.wzrost produkcji beta-globuliny
Pytanie 24
24.Mężczyzna z tranzycją A na G w pozycji 376 w eksonie V genu dehydrogenazy glukozo-6-fosfatazy powodującą zmianę asparaginianu w asparaginę, ma wariant A G6PD i:
a.przekaże to na swoich synow
e.powinien unikać bobu i niektorych lekarstw
b.dostanie anemii hemolitycznej
d.ma 10% aktywności G6PD
c.ma normalną lub pełną aktywność G6PD
Pytanie 25
25.Choroba HbH jest rzadko spotykana u Afroamerykanow, ponieważ:
c.alfa- talasemia u nich jest zwykle spowodowana mutacją nonsensowną w genie alfa globiny
e.anemia sierpowokrwinkowa chroni przed HbH
d.(--/) allele alfa globiny są u nich rzadkie
(alfa/- ) allele alfa globiny są u nich rzadkie
a.alfa talasemia jest bardzo rzadka u Afroamerykanow
Pytanie 26
26.Kariotypowo ok para ma dziecko z Downem. Analiza: dziecko: 1,2,2; 1,2,2; 1,1,1; 1,2,2 mama: 2,2; 2,2; 1,1; 1,2; tata: 1,2; 1,1; 1,1; 1,1;
ojcowska mejoza II
matczyna mejoza I
matczyna mejoza II
ojcowska mejoza I
ojcowska mejoza I lub II
Pytanie 27
Zaburzenie powodujące osłabienie mięśniowe i epilepsję. Ktory model dziedziczenia najprawdopodobniejszy?
mitochondrialny
nie-genetyczny, najprawdopodobniej środowiskowy
autosomalny dominujący
sprzężony z X recesywny
autosomalny recesywny
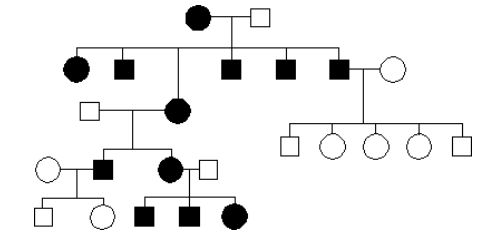
Pytanie 28
Ktore nie będzie dobrą metodą rozpoznania anemii sierpowokrwinkowej?
d.proba enzymatyczna aktwyności hemoglobiny
a.PCR eksonu 1 genu beta-globiny z następnym trawieniem restrykcyjnym MstII
b.trawienie restryktazami MstII genomowego DNA, następnie Southern blot, z użyciem produktu genu beta-globiny
e.elektroforeza hemoglobiny
c.PCR exonu 1 genu beta-globiny, następnie hybrydyzacja ASO oligonukleotydami, specyficznymi dla normalnych i nie alleli beta
Pytanie 29
Krotkie ramiona akrocentrycznych (satelity) chromosomow zawierają głownie wiele kopi genu kodującego rRNA
prawda
fałsz
Pytanie 30
30.Odkryte funkcje białek kodowanych przez protoonkogeny to wszystkie poza:
d.element szlaku transdukcji sygnału
a.czynnik wzrostu
c.enzym uczestniczący w naprawie zmismaczowanego DNA
b.receptor czynnika wzrostu
e.czynnik transkrypcyjny
Pytanie 31
31.Fenotypowo ok kobieta z kariotypem 45, XX, -14, -21, +t(14q, 21q) ma kariotypowo normalnego męża (46, XY). Pośrod ich żywego potomstwa najbardziej prawdopodobne są kariotypy 46, XX i 46, XY. A następne najbardziej prawdopodobne?
a.47, XY +21
b.46, XY, -14, +t(14q, 21q)
c.46, XY, -21, +t(14q, 21q)
e.45 XY, -14, -21, +16
d.45, XY, -14, -21, +t(14q, 21q)
Pytanie 32
Wyniki analizy odległości dla markera A i genu NF-1. Jaka jest odległość genetyczna między nimi?
0,5cM
30cM
10cM
0,1cM
1cM
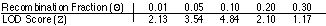
Pytanie 33
mitochondrialne
b.autosomalne recesywne
c.sprzężone z X recesywne
a.autosomalne dominujące
d.sprzężone z X dominujące
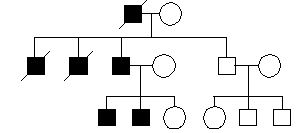
Pytanie 34
33.Młoda kobieta pochodzenia połnocnoeuropejskiego jest samotną matką dziecka z mukowiscydozą. Żeni się z innym połnocnoeuropejczykiem. Jakie jest prawdopodobieństwo, że on jest nosicielem mukowiscydozy? częstość 1/2500
1/1250
1/2500
1/25
1/50
1/4
Pytanie 35
Przedstawiona rodzina cierpi na zespoł Ehler-Danlos (zła synteza kolegenu- megarozciągliwość). Zaklasyfikowano każdego członka rodziny innym polimorficznym markerem DNA i wyszło to drzewko.
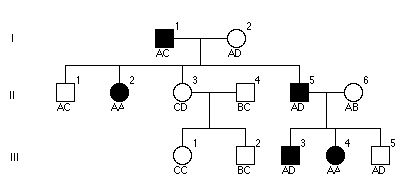
Pytanie 36
poniżej ukazany jest RFPL. „E” oznacza niezmienione miejsce restrykcji EcoRI. „*” polimorficzne miejsce EcoRI. Czarny pasek lokalizację sondy „A” DNA. Jakie są wszystkie możliwe allele (czyli długości paskow) możliwe do zobaczenia w Southern blot sondowanej przez „A”
d.2kb,3kb,6kb
c.3kb,4kb,6kb
b.1kb,3kb,4kb,6kb
e.żadne z powyższych
a.1kb,2kb,3kb,4kb,5kb,6kb
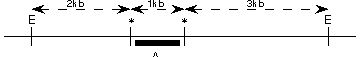
Pytanie 37
37.Ktora z poniższych chorob jest najmniej prawdopodobnie spowodowana aneuploidią?
b.Zespoł Turnera
d.Zespoł łamliwego chromosomu X
c.Zespoł Downa
a.Zespoł Klinefeltera
Pytanie 38
Przedsymptomowa diagnoza choroby Huntingtona wymaga analizy DNA przynajmniej 1 chorego członka rodziny
TAK
NIE
Pytanie 39
39.Ktora z poniższych fenotypowych kobiet ma rozwinięte jądra?
d.46, XY, ze sprzężonym z chromosomem X niedoborem receptora androgenow
c.46, XY, ze środmiąższową (?) delecją Yp włączając gen SRY
a.45, X
e.46, XX
b.46, XY, z mutacją punktową w domenie HMG w genie SRY
Pytanie 40
40.Para po 30 z 2 zdrowych dzieci poroniła dziewczynkę z wieloma wadami wrodzonymi. O czym powinniśmy rozmawiać z rodzicami zaraz po? CHYBA O CZYM NIE POWINNIŚMY XDDDD
b.Szansach zobaczenia, spędzenia czasu, potrzymania ich martwego dziecka
e.Możliwym doradztwie genetycznym
a.Dalszych badaniach dziecka (genetyczne, patomorfologiczne itp.) w celu wykrycia źrodła wad
c.Zachęcać matkę do natychmiastowego poddania się sterylizacji, żeby zapobiec sytuacji w przyszłości, zwłaszcza, ze ma już 2 zdrowych dzieci
d.Możliwości dotyczące pogrzebu itp.
Pytanie 41
41.Rodowod rodziny z zaburzeniami krzepinięcia. dziedzczenie:
b.sprzężone z X recesywne
e.sprzężone z X dominujące
d.autosomalne dominujące
c.mitochondrialne
a.autosomalne recesywne
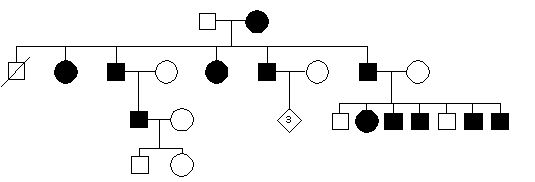
Pytanie 42
42.Ciężka beta-talasemia może nie ujawnić się klinicznie do kilku miesięcy po porodzie ponieważ:
b.noworodek ma minimalne wymagania tlenowe
d.zastąpienie gamma globiny beta globiną trwa do kilku miesięcy po porodzie
e.geny alfa globiny nie uaktywaniają się do kilku miesięcy po porodzie
c.podniesiony poziom HbA2 kompensuje brak genu beta globiny
a.Nadprodukcja zeta-globuliny kompensuje brakujący łańcuch beta
Pytanie 43
43.Ktore z poniższych nie może pojawić się jednocześnie u jednej osoby?
a.beta-talasemia i anemia sierpowokrwinkowa
b.alfa-talasemia i anemia sierpowokrwinkowa z genotypem SS
e.alfa-talasemia i beta-talasemia
d.5 kopi genu alfa globuliny (alfa,alfa, alfa/alfa, alfa)
c.alfa-talasemia i anemia sierpowokrwinkowa
Pytanie 44
44.Kobieta z bratem chorym autosomalnie recesywnie (100%penetracji podczas urodzenia) zrobiła test na nosicielstwo (98% czułości i 5% fałszywie dodatni). Wyszedł dodatni. Ryzyko bycia przez nią nosicielką wynosi:
50%
100%
33,3%
97,5%
66,7%
Pytanie 45
45.Nierowny crossing-over pomiędzy dwoma powtorzeniami Alu może doprowadzić do delecji lub duplikacji genu receptora LDL. Prawdą musi być:
a.gen receptora LDL zawiera tylko 1 sekwencję powtarzalną Alu
b.gen receptora LDL nie zawiera sekwencji powtarzalnych Alu
e.gen receptora LDL to sekwencja powtarzalna Alu
c.gen receptora LDL zawiera przynajmniej 2 sekwencje powtarzalne Alu
d.genom zawiera tylko jedną kopię sekwencji powtarzalnej Alu
Pytanie 46
46.Światowe rozmieszczenie braku beta-talasemi, anemii sierpowatej, G6PD jest związane z:
nietolerancją laktozy
grypą
stwardnieniem rozsianym
cholerą
malarią
Pytanie 47
Każdy rybonukleotyd zawiera dodatkową grupę 4’ OH, ktorej nie ma w DNA
PRAWDA
FAŁSZ
Pytanie 48
markery G8 RFLP są związane z locus choroby Huntingtona i są przydatne w analizie chorych rodzin. Podstawowe zmiany odpowiedzialne za polimorfizm G8 są rownież odpowiedzialne za Huntingtona.
TAK
NIE
Pytanie 49
49.„powtarzalny triplet” w chorobie Huntingtona to:
c.powtarzające się aminokwasy CAG
b.powtarzające się aminokwasy – Gly-X-Y
a.powtarzające się kwasy nukleinowe GAT
e.powtarzające się kwasy nukleinowe TAG
d.powtarzające się kwasy nukelinowe CAG
Pytanie 50
50.Średnie ryzyko powtorzenia się rozszczepienia wargi i podniebienia u dziecka (wieloczynnikowego defektu) to 4%. Jakie ryzyko wystąpi u pary, ktora ma 2 chorych dzieci?
c.10%
a.2%
d.25%
e.50%
b.4%
Pytanie 51
51.Dystrofia mięśniowa objawiać się może coraz większym nasileniem i dawać objawy coraz wcześniej w następujących po sobie pokoleniach. To zjawisko to:
c.składnik heterozygotyczny
a.antycypacja
d.niecałkowita penetracja
b.zrożnicowana ekspresja
e.heterogeniczność locus
Pytanie 52
Translokacja robertsonowska zazwyczaj dotyczy chromosomow
akrocentrycznych
metacentrycznych
Pytanie 53
Na podstawie rodowodu oceń jakie jest prawdopodobieństwo, że płod (romb) będzie miał dystrofię mięśniową Duchenne (choroba sprzężona z X recesywna)
1/36
1/18
1/4
1/8
1/9
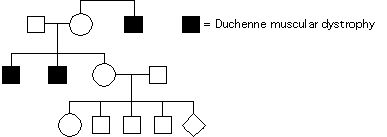
Pytanie 54
Poniżej ukazano rodowod rodziny z centralnej Słowacji z alkaptonurią. Model dziedziczenia to:
e.autosomalna recesywna
b.mitochondrialna
a.sprzężona z X recesywna
d.sprzężona z X dominująca
c.autosomalna dominująca
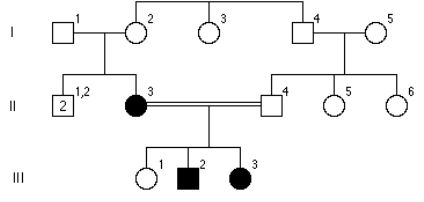
Pytanie 55
Jeżeli choroba X jest związana z markerem miejsca Y to musi istnieć związek między chorobą X, a allelem w miejscu Y.
55.NIE, bo związanie (?) jest mniej wiążące od sprzężenia.
Pytanie 56
56.Mężczyzna cichy nosiciel alfa talasemii (aa/a-) hajtnął się z kobietą z objawami alfa talasemi (aa/--) Zakładając, że nie dojdzie do rekombinacji ktory z genotypow nie jest możliwy u potomstwa?
a-/--
a-/a-
aa/a-
aa/aa
aa/--
Pytanie 57
56.42 letnia kobieta i jej 36 letni brat mają 10-15% aktywność alfa-1 antytrypsyny. Ona jest zdrowa, ale brat ma chorobę płuc. Zrożnicowanie ich stanu zdrowia jest najprawdopodobniej spowodowany:
e.wystawieniem na rożne warunku środowiskowe przez ostatnie 20 lat
a.genetyczną heterogenicznością
c.rożnymi allelicznymi wariantami tej choroby
d.posiadaniem rożnych prenatalnych warunkow środowiskowych
b.faktem, że ona ma 2 chromosomy X
Pytanie 58
Para (I-1, I-2) zbadała się genetycznie. Używając 2 par alellospecyficznych oligonukleotydow, jednej pary dla allelu BS (tzn jeden oligonukleotyd dla BS i drugi dla zdrowego BA) oraz para oligonukleotydow dla B+ allelu. Uzyskano wyniki: 58.Jakie jest prawdopodobieństwo, że płod (II-1) nie jest nosicielem ani beta-s, ani beta+?
1/2
1/4
3/4
1
0
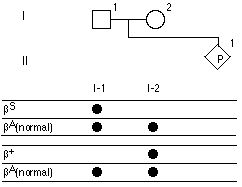
Pytanie 59
57.Para (I-1, I-2) zbadała się genetycznie. Używając 2 par alellospecyficznych oligonukleotydow, jednej pary dla allelu BS (tzn jeden oligonukleotyd dla BS i drugi dla zdrowego BA) oraz para oligonukleotydow dla B+ allelu. 59.Jakie jest prawdopodobieństwo, że płod będzie homozygotą *beta-s) z anemią sierpowatą?Uzyskano wyniki:
1/2
1
3/4
1/4
0
Pytanie 60
Kiedy dziecko będzie mieć 3 lata przeprowadzisz elektroforezę (dane poniżej). Jaki jest genotyp dziecka? HbA – 18%; HbS – 65%, HbF – 12%, HbA2-5%
Bs/B+
B/B
Bs/B
Bs/Bs
B+/B
Pytanie 61
61.Pacjenci z rodzinnym siatkowczakiem mają mutację w jednym genie Rb. Potencjalny mechanizm aktywacji drugiego allelu Rb u takich pacjentow to:
d.wszystkie powyższe
b.mitotyczny crossing-over
a.utrata normalnego chromosomu 13
c.niezależna mutacja punktowa
Pytanie 62
62.Typ III kolagenu jest homotrimerem łańcuchow alfa1 (III). Jeśli jeden z dwoch genow COL3A1 (kodujących to białko) zawiera mutację utraty sensu, ktora prowadzi do produkcji zmutowanego (lecz stabilnego) białka, jaki ułamek kolagenu typu III zawiera zmutowaną podjednostkę?
1/8
7/8
1/4
3/4
1/2
Pytanie 63
63.26 letni student medycyny bez problemow psychicznych i neurologicznych przebadał się pod kątem posiadania genu choroby Huntingtona (HD). Kilka osob w jego rodzinie łącznie z matką miało HD. Jego wyniki: HD allel 1: 17 powtorzeń; HD allel 2: 43 powtorzenia; najlepsza interpretacja to:
c.Jest bezobjawową jednostką, ktora odziedziczyła HD i u ktorej pojawią się objawy jeśli będzie żyć wystarczająco długo
b.odziedziczył 1 allel zmutowanego genu, ale biorąc pod uwagę brak symptomow choroby , najprawdopodobniej choroba się u niego nie rozwinie
e.wyniki są nierozstrzygające dlatego jest 50% szansy, że odziedziczona choroba nie została zauważona w teście
d.Ma HD i powinien poważnie rozważyć zmianę zawodu, bo nigdy nie będzie mogł kompetentnie praktykować
a. Na szczęście ma 1 normalny allel, więc jest tylko nosicielem, lecz nie będzie chory
Pytanie 64
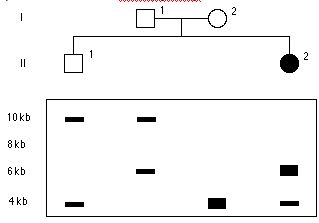
DNA ukazane poniżej jest z 3’ końcowego genu beta-globiny , ktory jest zmutowany w anemię sierpowatą. Ktore prążki będą widoczne w Southern blot od normalnego porbanta, strawione EcoRI i zhybrydyzowane z sondą A?
e.tylko prążki 6kb
b.tylko prążki 4kb
a.prążki 6kb i 10kb
c.prążki 4kb, 6kb, 10kb
d.prążki 4kb, 10kb
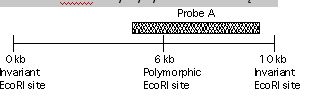
Pytanie 65
65.Ktora z poniższych obserwacji jest najmocniejszym dowodem na istotność czynnika genetycznego w powodowaniu cukrzycy typu 1
a.przeciwciała komorek Beta trzustki są często obecne
e.wspołczynnik konkordacji u bliźniąt jednojajowych jest 5 razy wyższy niż u dwu jajowych
d.wspołczynnik konkordacji u bliźniąt jednojajowych to 30%
c.choroba ujawnia się najczęściej w dzieciństwie
b.około 10% chorych ma chore rodzeństwo
Pytanie 66
66.Połnocnoeuropejka jest samotnym rodzicem dziecka z autosomalną recesywną mukowiscydozą. Ożeniła się z kuzynem I stopnia i zaszła w ciążę. Jakie jest prawdopodobieństwo, że jej dziecko będzie mieć mukowiscydozę?
1/32
1/2500
1/4
1/8
1/100
Pytanie 67
67.Wszystkie poniższe to mutacje zaobserwowane w allelach beta-talasemi oprocz:
Mutacja w incjatorze ATG
mutacna w AATAAAA poliA
mutacja G na A w początkowym GT w sekwencji donorowej splicingu
Mutacja zmiany sensu w kodonie 6 genu beta-globiny, zmieniające Glu na Val
mutacja tworząca nowe akceptory splicingowe w IVS-1
Pytanie 68
68.Zakładając rownanie Hardyego-Weinberga dla alleli mukowiscydozy w Kaukazkiej populacji i częstość zmutowanego allelu q=1/50 jaki procent ten populacji jest nosicielem mukowiscydozy?
1/100
(49/50)^2
2/50
(1/50)^2
1/50
Pytanie 69
U dużego odsetka chorych z niedoborem alfa1-antytrypsyny rozwinie się POChP albo rozedma. Choroba będzie miałą cięższy przebieg jeśli pacjent jest:
e.homozygotą bez alleli genu elastazy
d.heterozygotą posiadającą 1 prawidłową kopię genu alfa1- anytrypsyny
c.palaczem
a.kobietą
b. neutropenikiem
Pytanie 70
70.Każdy normalny łańcuch polipeptydowy wchodzący w kład kolagenu charakteryzuje się:
a.tripletem powtarzających się sekwencji aminokwasowych Cys-X-Y
b.kolejnymi powtorzeniami ALU
c.powtarzającym się tripletem sekwencji nukleotydow G-X-Y
d.powtarzającym się tripletem sekwencji nukelotydow C-A-G
e.powtarzającym się tripletem sekwencji aminokwasowych Gly-X-Y
Pytanie 71
71.Głowna forma hemoglobiny akumulująca się w płodzie z ciężką formą alfa-talasemi (obrzęk uogolniony płodu) składa się z:
2 delta + 2 beta
4 gamma
2 dzeta + 2 epsilon
4 alfa
4 beta
Pytanie 72
72.Mutacja nonsensowna w eksonie 14 skutkująca porażką receptora LDL w dotarciu do powierzchni komorki została odkryta w osobnikach z rodzinną hipercholesterolemią w wielu Libańskich rodzinach chrześcijańskich. Co wyjaśnia częstość występowania?
e.Libańczycy stosują dietę bogatą w tłuszcze zwierzęce
d.homozygoty mają przewagę przetrwaniową
c.efekt założyciela
a.duże rodziny
b.mutacja nie ma szkodliwych skutkowe przez co nie została odselekcjonowana
Pytanie 73
Rodzina przedstawiona poniżej cierpi na Dystrofię Mięśniową Duchenna, letalną, sprzężoną z chromosomem X. Osobnik III-1 ma 5 lat, osłabienie mięśniowe i nieprawidłową biopsję mięśni. Zarowno III-1 i III-2 (1 rok). Jakie jest ryzyko, że III-2 jest nosicielką?
1
3/4
1/4
1/2
0

Pytanie 74
74.Pacjent z anemią sierpowatą:
a.Ma inne mutacje w każdej kopii genu beta-globiny, ale obie są w tym samym kodonie
b.wykazuje nadmiar embrionalnej łańcuchow zeta w swoich dorosłych krwinkach
d.często umiera w macicy z powodu komplikacji związanych z hemoglobinopatią
e.Nie może mieć dziecka z anemią sierpowatą
c.nigdy nie doświadczył ataku choroby
Pytanie 75
75.Średnio jaki procent ciąż skutkuje w urodzeniu dziecka z poważną chorobą genetyczną lub defektem?
25%
0.1%
0%
3%
0.01%
Pytanie 76
76.Aby ustanowić z powodzeniem program badań przesiewowych w celu wykrycia heterozygot będących nosicielami chorob autosomalnych recesywnych wszystkie z poniższych są istotne oprocz:
a.program przesiewowy musi mieć 100% wartość przewidywania
e.dostępne są możliwości reprodukcyjne
b.doradztwo genetyczne jest zapewnione razem z testami
c.choroba jest na tyle poważna, żeby być istotną medycznie
d.populacja wysokiego ryzyka może być zidentyfikowana
Pytanie 77
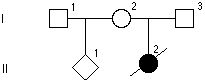
Rodowod poniżej przedstawia rodzinę z mukowiscydozą – chorobą autosomalną recesywną. Jakie jest prawdopodobieństwo, że I-3 jest nosicielem?
2/3
3/4
1/2
1/3
1/4
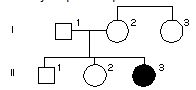
Pytanie 78
Częstość nosicielstwa choroby sprzężonej z X recesywnej o częstości występowania 1/10000 jest większe niż częstość nosicielstwa choroby autosomalnej recesywnej występującej z częstością 1/1000000.
tak
nie
Pytanie 79
Poniższa rodzina posiada allel dla choroby autosomalnej recesywnej 100% penetrującej. Pierwsza żona II-2 urodziła chore dziecko, ale umarła w połogu. Potem II-2 hajtnął się z przyrodnią siostrą II-2 (II-3), ktora zaszła w ciążę. Jaka jest szansa, że dziecko będzie chore?
1/4
5/32
1/2
q^2
1/16
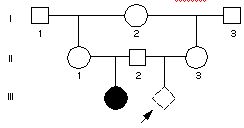
Pytanie 80
80.W jakich okolicznościach tajemnica lekarska może zostać złamana?
c.Pacjent psychiatryczny w przychodni mowi mi, że ma myśli o zamorodowaniu żony i teraz obmyśla jak to zrobić
b.23 letni pacjent zaczyna nową pracę i jego pracodawca rząda wynikow testu na HD
e. A i C
a.Młoda kobieta ma pozytywny wynik HIV, błaga, żeby nie mowić jej chłopakowi
d.Wszystkie powyższe
Pytanie 81
Probant III-1 ma mukowiscydozę. Analiza DNA wykazała, że III-1 jest homozygotą chorą, ale jego babcia I-2 jest homozygotą zdrową. q=1/50
Jakie jest prawdopodobieństwo, że I-1 jest nosicielem?
100%
(bez pary większe)
50%
Jakie jest prawdopodobieństwo, że III-2 jest chory?
0.5%
(bez pary mniejsze)
2%
jakie jest prawdopodobieństwo, że II-5 jest nosicielem?
4%
Pytanie 82
82.Anemia sierpowata:
a.jest spowodowana mutacją albo genu alfa globiny albo beta globiny
e.jest chorobą sprzężoną z X recesywną
b.rzadko prowadzi do tej samej mutacji u niespokrewnionych chorych
d.jest zawsze spowodowana tę samą mutacją punktową w genie beta globiny
c.jest chorobą autosomalną dominującą
Pytanie 83
83.26 latka pochodzenia Norweskiego zbadała się genetycznie. Jej brat umarł w wieku 8 lat na mukowiscydozę. Jej oboje rodzice zmarli. Kobieta przeszła testy DNA na 70 mutacji powodujących mukowiscydozę, ktore wykazują, że 90% nosicielow mukowiscydozy jest pochodzenia Europejskiego. Testy wyszły negatywnie na wszystkie 70 mutacji. Jakie jest prawdopodobieństwo, ze kobieta jest heterozygotyczną nosicielką?
poniżej 1%
2/3
1/15
1/6
1/25
Pytanie 84
Podczas inaktywacji chormosomu X, wszystkie geny na choromosmie X ulegają inaktywacji
NIE
TAK
Pytanie 85
W przypadku kiedy kobieta ma jeden chromosom X strukturalnie ok, a drugi nieok, chromosom ok. jest zazwyczaj inaktywowany.
NIE
TAK
Pytanie 86
86.„Jeden gen, jeden enzym” to zdanie obrazujące koncepcję:
d.zmiany w genach przez rzadkie enzymy są głowną przyczyną chorob metabolicznych
c.tylko 1 kopia genu musi być nieaktywna, żeby spowodować poważne dolegliwości związane ze spadkiem aktywności enzymatycznej poniżej 50%
e.większość chorob metabolicznych jest sprzężona z chromosomem X
a.poszczegolne etapy metaboliczne są kontrolowane przez poszczegolne enzymy kodowane przez specyficzny gen
b.enzymy funkcjonalne nigdy nie są złożone z podjednostek kodowanych przez rożne geny
Pytanie 87
Choroba Huntigtona jest spowodowana ekspansją powtorzeń trojek nukleotydow w
intronie 6
eksonie 6
Pytanie 88
88.Podejrzewałbyś możliwe zaburzenia kolagenu u pacjenta ktory ma jakiekolwiek z poniższych objawow oprocz:
e.postępujące pogorszenie neurologiczne
c.hipergiętkość stawow
d.niebieska twardowka
b.perforacje przewodu pokarmowego
a.wiele złamań
Pytanie 89
89.Ktore z poniższych z najmniejszym prawdopodobieństwem skutkuje allelem B0 dla beta-talasemia?
c.mutacja zmiany sensu wcześnie w eksonie 1 genu beta globiny
b.7bp delecja w ekosnie 2 genu beta globiny
e.delecja całego genu beta globiny
a.20bp insercja w eksonie 1 genu beta globiny
d.mutacja nonsensowna wcześnie w eksonie 2 genu beta globiny
Pytanie 90
90.Widzisz poprzednio zdrowego 3-latka obecnie bardzo ospałego, wymiotującego i z napadem. Ktory test laboratoryjny będzie najodpowiedniejszy, żeby potwierdzić podejrzenia?
c.poziom amoniaku w surowicy
b.tylko poziom fenyloalaniny w osoczu
d.24 godzinna kontrola poziomu kwasu homogentystycznego w moczu
e.sekwencja genu transkarbamylazy ornitynowej
a.poziom fenyloalaniny i tyrozyny w osoczu
Pytanie 91
91.Ktore zdanie jest zapowiedzią wieloczynnikowego modelu progowego?
a.odrębne ryzyko krewnych chorego wzrasta gdy powszechność chrooby spada
b.chore jednostki prawie zawsze mają chorego rodzica
d.wiek ujawnienia choroby spada, gdy byli chorzy krewni w 2 poprzednich pokoleniach
c.ryzyko jednostki nie wzrasta wraz z ilością chorych krewnych pierwszego stopnia
Pytanie 92
92.Hemofilia A i hemofilia B mają prawie identyczne fenotypy, ale powstają w wyniku mutacji rożnych genow na chromosomie X. Jest to przykład:
c.rożnej ekspresji
e.podwojnej heterozygotyczności
d.heterozygotyczności locus
a.czynnika heterozygotyczności
b.allelicznej heterozygotyczności
Pytanie 93
93.Ktory z poniższych kariotypow znajdziemy w ludzkim nasieniu?
b.23, X
c.46, XX
e.żadne z powyższych
d.46, XY
a.22, Y
Pytanie 94
Normalni rodzice mają dziecko z Downem. Poniżej ukazano haplotyp oparty na polimorficznym markerze locus niedaleko centromeru chromosomu 21. Na podstawie tego określ w ktorym podziale mejotycznym nastąpiła nondysjunkcja.
d.matczyna mejoza II
e.matczyna mejoza I lub ojcowska mejoza I
b.ojcowska mejoza II
a.ojcowska mejoza I
c.matczyna mejoza I
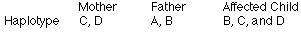
Pytanie 95
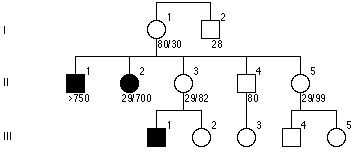
Zespoł łamliwego chromosomu X (FraX) jest chorobą sprzężoną z chromosomem X charakteryzującą się opoźnieniem umysłowym. Molekularną podstawą choroby jest ekspansja trpletow CGG w 1 eksonie genu FMR-1 na długim ramieniu chromosomu X. Zdrowi mają 6-52 powtorzeń, średnio 30, a chorzy >230 powtorzeń. Numerki w rodowodzie ot ilość powtorzeń. Jakie jest prawdopodobieństwo, że III-4 jest chory?
c.25%
e.80-100%
d.40-50%
a. poniżej 1%
b.10%
Pytanie 96
96.Zespoł Pradera Wiliego(PWS) może wynikać albo z delelcji środmiąższowej dotyczącej ojcowskiej kopi chromosomowego regionu 15q1-q13 albo od matczynej disomi jednorodzicielskiej chromosomu 15. Powodem tego jest:
a.PWS jest spowodowane anomalią w inaktywacji chromosomu X
b.Matczyna kopia gen(ow) odpowiedzialna za PWS nasila dominujący negatywny efekt ojcowskiego allelu
d.Matczyna kopia gen(ow) jest napiętnowana i nie ulega ekspresji
e.Ojczyna kopia gen(ow) jest napiętnowana i nie ulega ekspresji
c.matczyna kopia gen(ow) odpowiedzialna za zespoł Angelmana jest napiętnowana i nie ulega ekspresji
Pytanie 97
Choroba sprzężona z X recesywna, 100% penetracji. Jakie jest prawdopodobieństwo, że IV-5 będzie chorym synem?
1/8
1/4
1/18
1/36
1/9
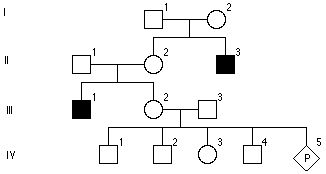
Pytanie 98
Rzadka choroba autosomalna, 100% penetracji. Choroba jest obecna i rozpoznawalna podczas urodzenia i nie zmniejsza sprawności. Założ brak heterogeniczności locus. Jaka jest szansa, że IV-1 jest chory?
4/9
1/9
1/4
1/2
2/9
Pytanie 99
99.Podczas dorosłego życia większość form hemoglobiny to:
2alfa + 2gamma
2alfa + 2delta
2zzeta + 2epsilon
2alfa + 2beta
2alfa + 2epsilon
Pytanie 100
100.HbH jest rzadko spotykana u potomkow Afroamerykanow, ponieważ:
a.hemoglobina Barta prawie zawsze towarzyszy genotypowi (a-/a-) u Afroamerykanow
c.alfa-talasemia występuje tylko w populacji azjatyckiej
b.chorzy na anemię sierpowatą wykazują odporność na efekty HbH
dominujący chromosom związany z alfa-talasemią u Afroamerykanow to (a-)
e.nadmiar tetramerow alfa2delta2 jest przyczyną HbH
Pytanie 101
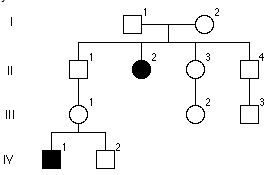
Rodowod rodziny z łamliwym chromosomem X. Ktora osoba najprawdopodobniej też jest chora?
a.III-1
d.IV-2
c.III-3
b.III-2
Pytanie 102
102.Wszystkie poniższe kariotypy są znajdowane w poronionych płodach. Ktory najmniej prawdopodobnie wystąpi u żywego dziecka?
a.46, XY
c.47, XX, +21
b.45, X
e.69, XXX
d.47, XX, +16
Pytanie 103
103.Doradztwo genetyczne obejmuje wszystkie za wyjątkiem:
b.dyskusję nt dostępnych testow genetycznych
c.dyskusję o wpływie choroby na pacjenta i rodzinę
e.zalecenie konkretnej metody reprodukcyjnej
a.ocena ryzyka pojawienia się choroby
d.dyskusję o dostępnych terapiach
Pytanie 104
Komorkowy fenotyp insercji lub delecji w domenie wiążącej ligand genu LDL-R to najczęściej:
a.niepowodzenie w syntezie każdego białka receptorowego LDL
b.niepowodzenie w transporcie receptora LDL przez ER
c.niepowodzenie rceptora LDL w zlokalizowaniu dołeczkow okrytych
e.niepowodzenie receptora LDL w byciu przyswojonym po związaniu LDL
d.niepowodzenie receptora LDL w związaniu LDL
Pytanie 105
105.Pomimo dużej ilości unikalnych mutacji w obrębie genu LDL-R, ktore powodują rodzinną hipercholesterolemię, niektore mutacje występują z dużą częstotliwością w niektorych populacjach np. 9,5kb delecja 3’ jest obserwowana u 35% Fińskich pacjentow. To przykład:
c.zrożnicowanej ekspresji
b.heterogeniczności locus
d.zbalansowanego polimorfizmu
a.efektu założyciela
e.allelicznej heterogeniczności
Pytanie 106
Rodzina z normalnym synem i corką z Downem. Zrobiono RFLP, oto wyniki. W ktorym podziale doszło do nondysjunkcji?
b.ojcowska mejoza II
d.matczyna mejoza II
a.ojcowska mejoza I
e.matczyna mejoza I lub matczyna mejoza II
c.matczyna mejoza I
Pytanie 107
107.Transplantacja wątroby może być skuteczna w leczeniu wszystkich poniższych zaburzeń za wyjątkiem:
d.ciężka rodzinna hipercholesterolemia
a.niedobor transkarbamylazy ornitynowej (OTC)
b.niedobor alfa1-antytrypsyny
c.niedobor dehydrogenazy glukozo-6-fosfatazowej (G6PD)
Pytanie 108
Zanalizowano sekwencję części genu Czynnika VIII u mężczyzny z hemofilią A i znaleziono mutację zmiany sensu zamieniającą Ile na Leu w kodonie 904. Analiza tego samego genu u innego niespokrewnionego wykazała taką samą zmianę. Drugi badany rownież musi mieć hemofilię A.
FAŁSZ
PRAWDA
Pytanie 109
109.G6PD jest popularny w Afrykańskich, Środziemnomorskich i Azjatyckich populacjach, w ktorych malaria jest endemiczna. Wiele rożnych mutacji zostało znalezionych w rożnych populacjach. Duża częstotliwość mutacji G6PD jest najlepiej wyjaśniona:
c.dryfem genetycznym
b.zrownoważonym polimorfizmem (dobor stabilizujący?)
e.łagodny fenotyp deficytu G6PD
d.efekt założyciela
a.wpływem lekow, ktore powodują anemię hemolityczną
Pytanie 110
Ktore z poniższych są charakterystycznymi klinicznymi objawami rodzinnego nowotworu?
a.Wcześniejszy rozwoj tego nowotworu w porownaniu ze sporadycznymi formami tego samego nowotworu
żadne
b.2 lub więcej niezależnych początkow raka u jednej osoby
c.rzadko widuje się te typy nowotworow jako sporadyczne nowotwory
a+b
a+b+c
Pytanie 111
111.Ktory z podanych kariotypow jest najczęściej spotykany u żywych urodzeń?
d.46, XY, -11, +21
e.69, XXX
a.47, XX, +3
c.46, YY
b.47, XX, +21
Pytanie 112
112.Zarowno typ I jak i typ II osteogenesis imperfecta (wrodzona łamliwość kości) jest dziedziczona autosomalnie dominująco i często spowodowana mutacją genu alfa1 (I) kolagenu. Większość mutacji powodujących łagodniejszy typ I to null (zerowe), podczas gdy te powodujące letalny typ II to najczęściej mutacje zmiany sensu, ktore umożliwiają syntezę normalnych ilości zmienionych podjednostek kolagenu. Wyjaśnieniem tego paradoksu jest to, że w typie II:
d.wszystkie cząsteczki prokolagenu są normalne, ale ich całościowa jakość jest zredukowana o 50%
c.50% cząsteczek prokolagenu jest wadliwych
a.100% cząsteczek prokolagenu jest wadliwych
b.75% cząsteczek prokolagenu jest wadliwych