Wyszukaj
Zaloguj / zarejestruj się
Jeśli masz już konto, możesz się zalogować.
Jeśli nie masz konta, zarejestruj nowe podając nazwę użytkownika, adres email i hasło.
Formularz kontaktowy
Memorizer+
Wykup dostęp
Ta funkcja jest dostępna dla użytkowników, którzy wykupili plan Memorizer+
Fiszki
Genetyka z Michigan XD
Test w formie fiszek GENETYKA XD
Ilość pytań: 112 Rozwiązywany: 3678 razy
21.Choroba autosomalna recesywna występuje z częstością 1/10000 w populacji (zakładamy, że w danej miejscowości jest 100k ludu). Kumpel twierdzi, że ma chorobę chociaż nie miał w rodzinie bo test wyszedł pozytywnie (98% wrażliwości, 90% specyficzności). Jaka jest szansa, że jednak nie ma?
10%
83%
17%
49%
2%
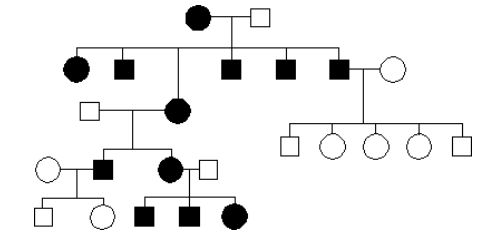
22.Jakie jest dziedziczenie w tym rodowodzie?
b.autosomalne dominujące
d.sprzężone z X recesywne
a.autosomalne recesywne
e.mitochondrialne
c.sprzężone z X dominujące

23.Ktore z poniższych nie pomoże pacjentowi z beta-talasemią major (najcięższa)
e.wzrost produkcji HbF
a.wzrost produkcji beta-globuliny
c.spadek produkcji alfa-globuliny
b.wzrost produkcji alfa-globuliny
d.transfuzja i chelatacja żelazem
24.Mężczyzna z tranzycją A na G w pozycji 376 w eksonie V genu dehydrogenazy glukozo-6-fosfatazy powodującą zmianę asparaginianu w asparaginę, ma wariant A G6PD i:
e.powinien unikać bobu i niektorych lekarstw
a.przekaże to na swoich synow
b.dostanie anemii hemolitycznej
c.ma normalną lub pełną aktywność G6PD
d.ma 10% aktywności G6PD
25.Choroba HbH jest rzadko spotykana u Afroamerykanow, ponieważ:
d.(--/) allele alfa globiny są u nich rzadkie
c.alfa- talasemia u nich jest zwykle spowodowana mutacją nonsensowną w genie alfa globiny
e.anemia sierpowokrwinkowa chroni przed HbH
(alfa/- ) allele alfa globiny są u nich rzadkie
a.alfa talasemia jest bardzo rzadka u Afroamerykanow
26.Kariotypowo ok para ma dziecko z Downem. Analiza: dziecko: 1,2,2; 1,2,2; 1,1,1; 1,2,2 mama: 2,2; 2,2; 1,1; 1,2; tata: 1,2; 1,1; 1,1; 1,1;
ojcowska mejoza I
ojcowska mejoza I lub II
matczyna mejoza I
ojcowska mejoza II
matczyna mejoza II
Zaburzenie powodujące osłabienie mięśniowe i epilepsję. Ktory model dziedziczenia najprawdopodobniejszy?
mitochondrialny
autosomalny recesywny
nie-genetyczny, najprawdopodobniej środowiskowy
autosomalny dominujący
sprzężony z X recesywny
Ktore nie będzie dobrą metodą rozpoznania anemii sierpowokrwinkowej?
a.PCR eksonu 1 genu beta-globiny z następnym trawieniem restrykcyjnym MstII
e.elektroforeza hemoglobiny
b.trawienie restryktazami MstII genomowego DNA, następnie Southern blot, z użyciem produktu genu beta-globiny
d.proba enzymatyczna aktwyności hemoglobiny
c.PCR exonu 1 genu beta-globiny, następnie hybrydyzacja ASO oligonukleotydami, specyficznymi dla normalnych i nie alleli beta
Krotkie ramiona akrocentrycznych (satelity) chromosomow zawierają głownie wiele kopi genu kodującego rRNA
fałsz
prawda
30.Odkryte funkcje białek kodowanych przez protoonkogeny to wszystkie poza:
a.czynnik wzrostu
c.enzym uczestniczący w naprawie zmismaczowanego DNA
b.receptor czynnika wzrostu
d.element szlaku transdukcji sygnału
e.czynnik transkrypcyjny
31.Fenotypowo ok kobieta z kariotypem 45, XX, -14, -21, +t(14q, 21q) ma kariotypowo normalnego męża (46, XY). Pośrod ich żywego potomstwa najbardziej prawdopodobne są kariotypy 46, XX i 46, XY. A następne najbardziej prawdopodobne?
b.46, XY, -14, +t(14q, 21q)
a.47, XY +21
d.45, XY, -14, -21, +t(14q, 21q)
e.45 XY, -14, -21, +16
c.46, XY, -21, +t(14q, 21q)
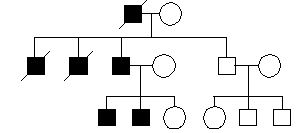
Wyniki analizy odległości dla markera A i genu NF-1. Jaka jest odległość genetyczna między nimi?
30cM
1cM
0,1cM
10cM
0,5cM
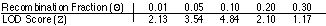
a.autosomalne dominujące
d.sprzężone z X dominujące
c.sprzężone z X recesywne
b.autosomalne recesywne
mitochondrialne
33.Młoda kobieta pochodzenia połnocnoeuropejskiego jest samotną matką dziecka z mukowiscydozą. Żeni się z innym połnocnoeuropejczykiem. Jakie jest prawdopodobieństwo, że on jest nosicielem mukowiscydozy? częstość 1/2500
1/1250
1/50
1/2500
1/4
1/25
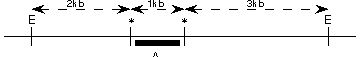
Przedstawiona rodzina cierpi na zespoł Ehler-Danlos (zła synteza kolegenu- megarozciągliwość). Zaklasyfikowano każdego członka rodziny innym polimorficznym markerem DNA i wyszło to drzewko.
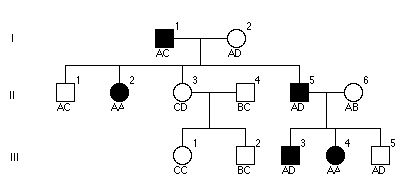
poniżej ukazany jest RFPL. „E” oznacza niezmienione miejsce restrykcji EcoRI. „*” polimorficzne miejsce EcoRI. Czarny pasek lokalizację sondy „A” DNA. Jakie są wszystkie możliwe allele (czyli długości paskow) możliwe do zobaczenia w Southern blot sondowanej przez „A”
d.2kb,3kb,6kb
a.1kb,2kb,3kb,4kb,5kb,6kb
e.żadne z powyższych
b.1kb,3kb,4kb,6kb
c.3kb,4kb,6kb
37.Ktora z poniższych chorob jest najmniej prawdopodobnie spowodowana aneuploidią?
b.Zespoł Turnera
c.Zespoł Downa
a.Zespoł Klinefeltera
d.Zespoł łamliwego chromosomu X
Przedsymptomowa diagnoza choroby Huntingtona wymaga analizy DNA przynajmniej 1 chorego członka rodziny
TAK
NIE
39.Ktora z poniższych fenotypowych kobiet ma rozwinięte jądra?
d.46, XY, ze sprzężonym z chromosomem X niedoborem receptora androgenow
a.45, X
e.46, XX
b.46, XY, z mutacją punktową w domenie HMG w genie SRY
c.46, XY, ze środmiąższową (?) delecją Yp włączając gen SRY
40.Para po 30 z 2 zdrowych dzieci poroniła dziewczynkę z wieloma wadami wrodzonymi. O czym powinniśmy rozmawiać z rodzicami zaraz po? CHYBA O CZYM NIE POWINNIŚMY XDDDD
e.Możliwym doradztwie genetycznym
b.Szansach zobaczenia, spędzenia czasu, potrzymania ich martwego dziecka
d.Możliwości dotyczące pogrzebu itp.
a.Dalszych badaniach dziecka (genetyczne, patomorfologiczne itp.) w celu wykrycia źrodła wad
c.Zachęcać matkę do natychmiastowego poddania się sterylizacji, żeby zapobiec sytuacji w przyszłości, zwłaszcza, ze ma już 2 zdrowych dzieci
Cześć!
Wykryliśmy, że blokujesz reklamy na naszej stronie.
Reklamy, jak zapewne wiesz, pozwalają na utrzymanie i rozwój serwisu. W związku z tym prosimy Cię o ich odblokowanie by móc kontynuować naukę.